



































































































































































































































Nature | 上海药物所破解GPCRs的激酶招募及偏向性信号转导机制
2023年8月2日,中国科学院上海药物研究所徐华强研究员、段佳研究员和杨德华研究员共同在Nature杂志上发表了最新研究成果“GPCR activation and GRK2 assembly by a biased intracellular agonist”,在国际上报道了第一个高分辨率GPCR——神经降压素受体(neurotensin receptor 1,NTSR1)与GRK2的复合物结构,揭示了GRK2识别和调控GPCR的详细分子机制,并通过结构解析,首次发现了一个全新的GPCR偏向性配体结合口袋,为临床开发靶向GPCR的偏向性药物分子开辟了全新的思路和途径。
G蛋白偶联受体(G Protein-Coupled Receptors, GPCRs)是一类广泛存在于人体细胞膜上的膜受体,是细胞信号转导的重要调节分子。GPCRs参与调控人体几乎所有的生命活动过程,从化学感知,包括视觉、嗅觉、味觉,到内分泌分子相关的调节,包括神经传递、免疫调节、代谢调节等。人体基因组能编码超过800个GPCRs。目前,FDA批准上市的临床药物中,约三分之一的药物作用于GPCRs发挥治疗作用,GPCRs被认为是新药研发领域中最重要,也是最有应用前景的药物靶点之一。
GPCRs在被配体激活后,主要通过下游的G蛋白或arrestin通路行使特定的生理功能。然而,GPCRs在招募arrestin蛋白之前,必须被GPCR激酶(GPCR Kinases,GRKs)识别和调控。GRKs能够磷酸化GPCRs,促进其招募arrestin蛋白,抑制其招募G蛋白,GRKs被认为是调控GPCR两条信号通路转换的关键分子。因此,在整个GPCR信号转导领域存在三个最为关键的科学问题,分别是:1)GPCRs如何识别和招募下游G蛋白;2)GPCRs如何识别和招募下游arrestin蛋白;3)GPCRs如何被GRKs识别和调控。
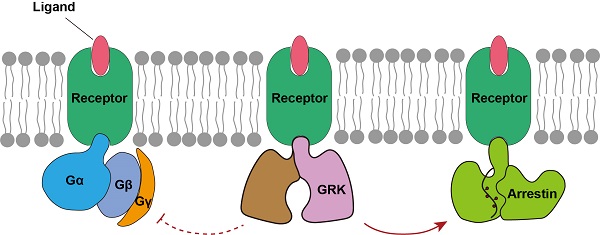
图1 GPCRs信号转导领域最关键的三个科学问题
2011年,斯坦福大学Brian K. Kobilka实验室解析了第一个GPCR——β肾上腺素能受体与Gs蛋白的复合物晶体结构,揭示了GPCR与G蛋白识别的分子基础 (Rasmussen et al., 2011)。Brian K. Kobilka也因在GPCR研究领域的突出贡献与他的博士导师Robert J. Lefkowitz共同获得了2012年的诺贝尔化学奖。2015年,徐华强课题组利用自由电子激光首次解析了人视紫红质蛋白rhodopsin与arrestin1的复合物结构,揭示了GPCR与arrestin蛋白识别的分子基础 (Kang et al., 2015),并在之后进一步解析了GPCRs招募arrestin蛋白的磷酸化密码 (Zhou et al., 2017)。然而,由于GPCRs与GRKs的结合短暂且高度动态,想要获得稳定的GPCR-GRK复合物非常困难,这使得有关GPCRs与GRKs的结构与功能研究进展缓慢。
早在十年前,徐华强课题组便展开了GPCR与GRK复合物的结构研究工作,然而,鉴于GPCR与GRK之间的微弱相互作用,阻碍了进一步的结构研究,因此,亟待建立新的技术手段或方法来克服复合物不稳定的难题。2017年,徐华强课题组和斯坦福大学Brian K. Kobilka实验室均通过借助大量的生化实验、分子模拟、细胞试验等提出了GPCR识别GRK的分子模型。直到2021年,普渡大学John J. G. Tesmer实验室在Nature上报道了第一个GPCR——视紫红质蛋白rhodopsin与GRK1的复合物冷冻电镜结构 (Chen et al., 2021),首次揭示了视觉GPCR识别GRK1的模式,然而,由于复合物整体密度较差,GRK1只有催化结构域能被确定,且受体与GRK1相互作用界面的分子基础仍不清晰。
另一方面,GPCRs作为一类重要的药物靶标,越来越多的研究发现,GPCR的两条信号通路,G蛋白和arrestin通路,与靶向GPCRs的药物分子的疗效和副作用密切相关。相比于传统的GPCR激动剂或抑制剂能够同时激活或抑制GPCR的两条信号通路,选择性激活(即偏向性激动剂)GPCR其中一条通路的药物分子往往具有更好的疗效和更低的副作用,因此,开发偏向性药物分子是靶向GPCR药物研发的重要趋势。然而,截至目前,绝大多数偏向性激动剂均是通过偶然筛选发现的,且作用机制不明确。GRKs是调控GPCR两条信号通路转换的关键分子,GRKs也被认为是靶向GPCR偏向性药物分子发现的关键一环。研究GRKs识别和调控GPCRs的分子机制,揭示偏向性激动剂介导GPCR的偏向性信号转导机制,对于靶向GPCR的偏向性药物发现具有十分重要的科学意义。
由于GPCRs与GRKs的微弱相互作用是阻碍GPCR-GRK复合物结构研究的最大难点。因此,在本次研究中,段佳研究员主要通过选择合适的研究对象、引入NTSR1偏向性激动剂SBI-553、引入NanoBiT交联技术、采用化学交联技术四种策略获得了稳定的GPCR-GRK复合物从而实现了结构解析。
本项研究是在徐华强课题组十年研究基础上,首次成功解析高分辨率的GPCR-GRK复合物结构,揭示了GPCR信号转导领域现存的一个最为关键的科学问题,即GPCRs如何受GRKs识别和调控,这是细胞信号转导里程碑式的成果。值得一提的是,徐华强课题组7月31日刚在Nature上报道了B类GPCRs的新型G蛋白偏向性小分子激动剂,揭示了G蛋白偏向性配体的作用机制,结合本项研究,两项研究首次全面系统地揭示了两种不同类型的偏向性激动剂介导受体偏向性信号转导的详细分子机制,极大地促进了我们对GPCRs偏向性信号转导的理解与认识,为今后开发靶向GPCRs的偏向性药物分子提供了夯实的结构基础。
上海药物所段佳研究员、刘恒博士、赵丰辉博士为本论文共同第一作者。上海药物所徐华强研究员、段佳研究员和杨德华研究员为共同通讯作者。上海市高峰电镜中心执行主任袁青宁对数据的收集及处理提供了大量帮助。该工作得到了国家重点研发计划、上海市市级科技重大专项、中国科学院战略性先导科技专项、国家自然科学基金委等项目的资助。
专家点评:
饶子和 中国科学院院士
结构是功能的基础。生物分子的独特三维构象决定了其分子行为,解析生物分子的三维结构对理解其生物学功能至关重要。GPCR是最大的细胞膜蛋白家族,在细胞通讯和众多生命过程中发挥关键作用,也是药物治疗或干预的主要目标,约三分之一的FDA批准上市的药物通过靶向GPCR发挥作用。
GPCR复合体的动态组装,涉及各种相互作用的蛋白质,如G蛋白、arrestin以及GPCR激酶(GRK),是其发挥功能的关键调节蛋白。GPCR信号转导复合体执行多种细胞过程,具有动态性和灵活性,这突显了生命系统的适应性,但同时也使得对它们的研究充满挑战。
尽管面临着巨大的挑战,理解GPCR及其相关复合体的结构是非常必要的。结构提供了对GPCR功能、信号转导机制以及如何利用这些相互作用进行药物治疗干预的详细的指导依据,为理性药物设计铺平了道路,促进了更具精确性和靶向性的创新药物开发。
GPCR与下游调节蛋白的相互作用瞬时且短暂,在三种类型的调节蛋白中,GPCR与G蛋白的亲和力最高,在纳摩尔级别,而与GRK的亲和力最低,低于微摩尔级别,这使得获得GPCR-GRK复合体的高分辨率结构在技术上比获得GPCR-G蛋白复合体、GPCR-arrestin复合体的高分辨率结构更具有挑战性。GPCR与GRK的弱相互作用使得使用X射线晶体学等传统技术进行研究变得无能为力,因此需要借助冷冻电子显微镜(cryo-EM)技术结合多种生化技术手段等开展研究。
GPCR与GRK之间的相互作用是GPCR信号传导的关键步骤。这篇论文代表了GPCR信号领域的重大突破,提供了神经递质受体NTSR1与GRK2和SBI-553之间相互作用的详细结构分析。该研究使用冷冻电子显微镜(cryo-EM)获得了与SBI-553结合的NTSR1-GRK2复合体的高分辨率结构,SBI-553是一种选择性激活arrestin通路的偏向性激动剂。该研究首次揭示了NTSR1和GRK2之间的详细相互作用,提供了SBI-553诱导NTSR1-GRK2相互作用的分子机制。值得提出的是,该项研究中揭示的SBI-553的结合口袋,位于受体靠近胞内侧的疏水口袋中,与此前发现的所有GPCR配体结合口袋的位置均不同,这为靶向GPCR的偏向性药物分子开发提供了全新的思路和途径。
徐华强课题组在研究动态的生物大分子复合物具有丰富的经验。十几年前,徐华强课题组就利用X射线晶体学率先解析了植物抗逆激素ABA受体复合物 (Nature 2009)和其关键信号传导激酶SnRK-PPC的动态复合物结构 (Science 2012)。 在2013年,徐华强课题组利用自由电子激光解析了第一个GPCR-arrestin结构 (Nature 2015),并通过结构解析,破解了GPCR磷酸化密码 (Cell 2017)。GPCR-GRK2课题更具有挑战性,是近年来GPCR信号转导领域遗留的最大的一个科学问题。徐华强/段佳课题组与杨德华课题组通力合作,通过进一步发展NanoBiT交联方法,并结合化学分子交联与冷冻电镜技术,攻克了GPCR-GRK2这一难题,这将是GPCR领域又一个里程碑式成果。我衷心祝贺徐华强,段佳,和杨德华团队,并期待他们的下一个突破性成果。
裴钢 中国科学院院士
G蛋白偶联受体(GPCRs)超家族是最大和功能最多的细胞表面受体群,能够响应各种外部刺激,从而介导不同的生理功能。人类基因组编码800多种GPCRs。GPCRs在细胞表面的可达性及其配体识别模式的多样性使得GPCRs成为最关键的药物靶点,大约三分之一的所有上市药物通过作用于GPCRs发挥作用。
GPCRs主要通过两条途径传导下游信号:G蛋白途径和休止蛋白(arrestin)途径。G蛋白途径在GPCR被配体刺激后被激活,GPCR促进G蛋白上的GDP与GTP的交换,导致G蛋白分离成α和βγ亚基。这些亚基可以进一步调控各种下游效应蛋白并介导不同通路。另一方面,arrestin途径涉及arrestin被招募到激活和被G蛋白偶联受体激酶(GRKs)磷酸化修饰的GPCR上,导致受体失活、内化和进一步的信号传导。
GPCRs药物发现中的一个重大挑战是理解和利用偏向性信号传导的概念。偏向性配体可以选择性地只激活其中一条信号途径,这为提高药物治疗效能和减少副作用提供了可能。然而,由于GPCR信号传导的复杂性和对偏向信号转导机制的理解不完全,设计偏向性配体是十分困难的。尽管存在这些挑战,寻求偏向性配体仍代表了药物发现国际竞争中令人兴奋的前沿,可能会带来更安全、更有效的治疗药物。
此外,揭示偏向性信号转导的结构基础充满挑战。GPCRs存在多种构象状态,不同的配体可以将受体稳定在不同的状态。同时,GPCRs、G蛋白和arrestin的蛋白-蛋白相互作用的瞬态性质进一步复杂化了这些研究。在生物学相关的环境中捕获GPCRs的结构动力学并将这些动力学与偏向性信号传导联系起来仍然是一个重大难题。除了GPCRs本身的复杂性外,理解它们与G蛋白偶联受体激酶(GRKs)的交互也是具有挑战性的。GRKs对GPCR信号传导至关重要,GRKs负责磷酸化激活的GPCRs,从而促进GPCRs招募arrestin,导致受体失活、内化和进一步的信号传导。这种交互的瞬态和动态性质,使得GPCR-GRK复合物的结构表征是十分困难的。目前,尚未报道完整的GPCR与GRK复合物的高分辨率结构,更加突显了GPCR-GRK结构解析充满技术挑战。然而,GPCR-GRK结构解析将极大地促进理解GPCR的功能调控机制,并在设计更具针对性的治疗策略方面提供重要的指导依据。
这篇论文的研究成果是GPCR信号转导领域又一个重大突破,并有可能改进药物开发从而改善患者的治疗疗效。该研究提供了神经降压素受体NTSR1、GRK2和偏向性配体SBI-553之间互作的详细结构分析,揭示了GPCR信号转导和偏向性激动作用的分子机制。这种近原子水平的结构信息以前从未报道过,为开发更精确、效果更好地针对GPCR的新药物奠定了结构基础。该研究突出了GRK2在GPCR激活和偏向性信号中的重要性,展示了偏向性激动作用作为GPCR相关疾病的治疗策略的潜力,通过理解偏向性激动剂的分子机制,研究者可以设计出选择性地只激活G蛋白或arrestin途径的药物,从而获得更好的治疗效果和安全性的药物。这对开发治疗一系列疾病,包括癌症、心血管疾病和神经系统疾病等的创新药物具有重大意义。
徐华强实验室长期致力于GPCR结构生物学和药物设计,其实验室解析了第一个GPCR-arrestin复合物结构(Nature 2015)并破解了GPCR磷酸化密码(Cell 2017)。NTSR-GRK2的复合物结构是GPCR信号转导领域的又一个重大突破,该项研究中所建立的方法和技术将对今后解析其他GPCR动态复合物结构具有重要的指导意义。
(供稿部门:徐华强课题组;供稿人:段佳)