





























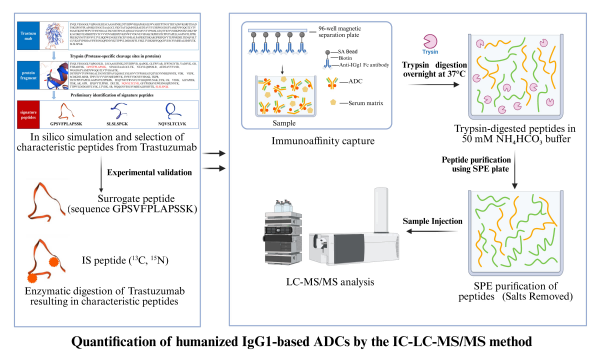
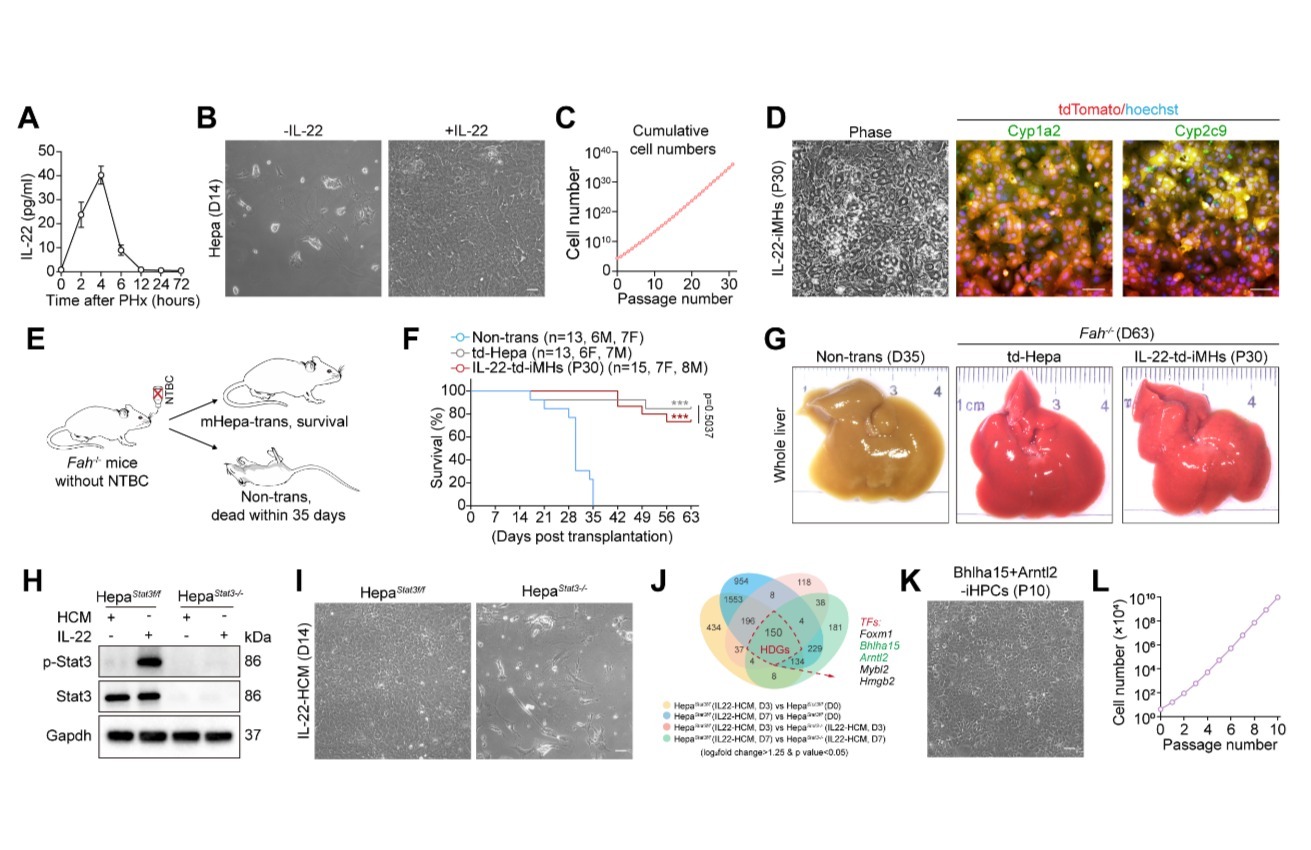
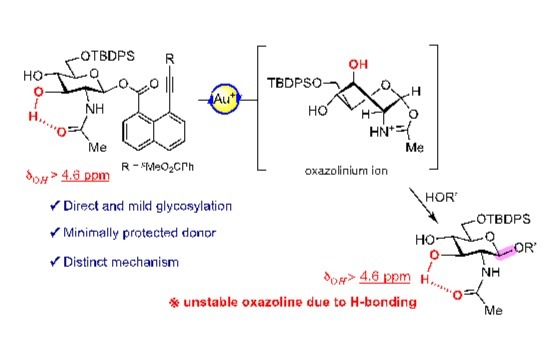

































































































































































































Nat Comput Sci | PBCNet:药物先导化合物优化的人工智能新方法
先导化合物的结构优化(Lead optimization)是药物设计的核心环节,需要通过DMTA(设计-合成-测试-分析)的反复循环来提高化合物活性、特异性、成药性等性质。长期以来,先导化合物的活性优化高度依赖药物化学家的经验以及大量的人力和资源投入。基于结构的药物设计,尤其是高精度的结合自由能计算,可以通过部分模拟的DMTA循环来加速先导化合物活性优化过程。随着分子力场和构象采样算法的改进,自由能微扰(FEP)等相对结合自由能模拟方法的预测值与实验结果的误差可以接近化学精度(1 kcal/mol,约5-6倍活性差异以内),但这类方法通常需要复杂的配置与体系搭建过程。此外,消耗计算资源庞大、商业软件价格高昂等问题也限制了这类方法的应用范围。因此,开发一种兼顾速度、精度与易用性的先导化合物活性优化方法一直是药物设计领域的迫切需求。
近期,中国科学院上海药物研究所郑明月课题组提出了一种先导化合物优化的人工智能方法PBCNet(pairwise binding comparison network)。该方法采用孪生图卷积神经网络架构,通过比较一组相似配体的结合模式差异来预测二者之间的相对结合亲和力,可以较好的兼顾计算速度和精度。此外,研究团队还开发了易于操作的图形界面网络计算服务(https://pbcnet.alphama.com.cn/index)。相关研究论文“Computing Relative Binding Affinity of Ligands Based on a Pairwise Binding Comparison Network”于2023年10月19日于Nature Computational Science在线发表。
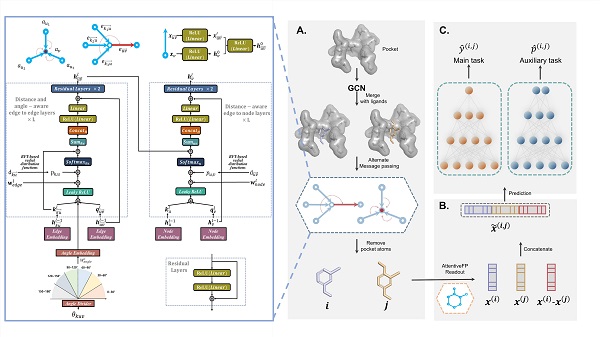
图1 PBCNet网络主体框架。A. 信息传递阶段,实现配体与蛋白质分子之间相互作用的信息交互,并获得配体原子级别的表征信息;B. 读出阶段,获取配体分子级别的隐空间表征,并实现成对配体之间的信息交互;C. 预测阶段,配体分子对表征经过两支独立的3层前馈神经网络,分别输出:(1)两个配体分子对在该蛋白体系下的 pIC_50预测值y(i,j);(2)第一个配体分子的结合亲和力高于第二个配体分子结合亲和力的预测概率p(i,j)。
近年来,人工智能技术已逐渐成为药物研发领域中重要的技术手段之一。基于已知的结构和活性数据,如何引入合理的关系归纳偏置使得AI模型可以更有效的提取其中蕴含的物理化学知识和分子相互作用规律,是开发先导化合物优化方法的关键。在PBCNet中,模型采用了一种孪生网络架构(图1),可以较好的消除不同来源或实验测定条件下结合亲和力数据中存在的系统误差;考虑到分子间非键相互作用往往服从严格的几何先验,模型将原子之间的距离和化学键之间的角度信息编码作为注意力偏置项,有助于分子间相互作用信息的交互;此外,相对于二维拓扑图,模型同时引入距离和角度信息得到三维分子图,可以更全面的描述了蛋白-配体之间的分子结合信息。
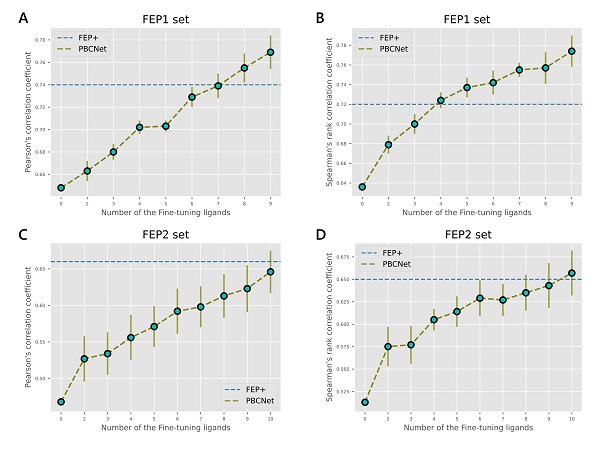
研究团队设计了多种测试场景,对包括PBCNet在内的多种先导化合物优化方法进行了性能比较。在零样本学习方面, PBCNet的排序能力和预测精度均显著优于Schr dinger Glide、MM-GB/SA和多个近期报道的深度学习模型,在部分测试体系上的误差已接近于化学精度;在小样本学习方面,仅使用少量的结构活性数据对模型进行微调后PBCNet的排序性能已经可以接近或超过Schr dinger FEP+(图2),并且在计算速度方面相对FEP+有多个数量级的优势。对比结果显示,使用PBCNet可以使先导化合物优化项目平均加速约4.7倍,平均节省约30%的资源投入。此外,模型在原子和官能团水平的可解释性分析也反映了PBCNet预测结果的合理性。
图2 小样本学习中PBCNet的性能随微调样本量(已知活性化合物数量)的变化趋势。x轴表示微调样本的数量,y轴表示PBCNet的预测性能,蓝色虚线为文献报道的Schr dinger FEP+的预测性能。误差线表示10次独立运行的标准偏差。
本论文的第一作者为上海药物所研究生虞杰和苏州阿尔脉生物科技有限公司的李召军博士。上海药物所郑明月研究员、罗小民研究员、博士后李叙潼为本研究论文的共同通讯作者。本研究得到了国家自然科学基金、临港实验室、国家重点研发专项、中国博士后科学基金、上海市自然科学基金、上海药物所与上海中医药大学中医药创新团队联合研究项目、以及上海市科技重大专项资助。
Research Briefing: https://doi.org/10.1038/s43588-023-00531-1
(供稿部门:郑明月课题组)